复合金属氢化物由于高容量、成本低、安全性好等因素是储氢材料的研究热点之一。然而这些材料的热力学的稳定好,解吸势垒高导致氢气解离温度高,氢气解离和吸附的反应速率慢。通过掺杂过渡金属(TM)来调控热力学和动力学性质是解决这些问题的有效手段。
本课题利用第一性原理和动力学模拟等多种手段来研究过渡金属掺杂复合金属氢化物的氢气解吸机理。主要研究成果包括确定掺杂金属和主体之间相互作用的微观结构、过渡金属在氢气解吸机理中的作用、氢气解吸的化学反应和物理相变的偶合机理等。
(1)微观结构和热力学性质
微观结构计算揭示了TM进入NaAlH4的表面结构的间隙位置,形成了TMAl3H12的微观结构,这个结构改变了NaAlH4的热力学稳定性,有效的促进了氢气的解离。实验测定验证了我们的计算预测。
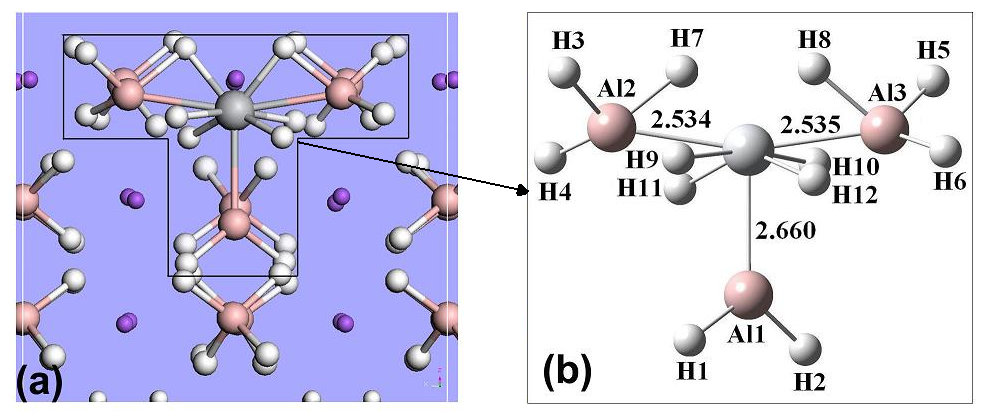
(2)电子结构分析
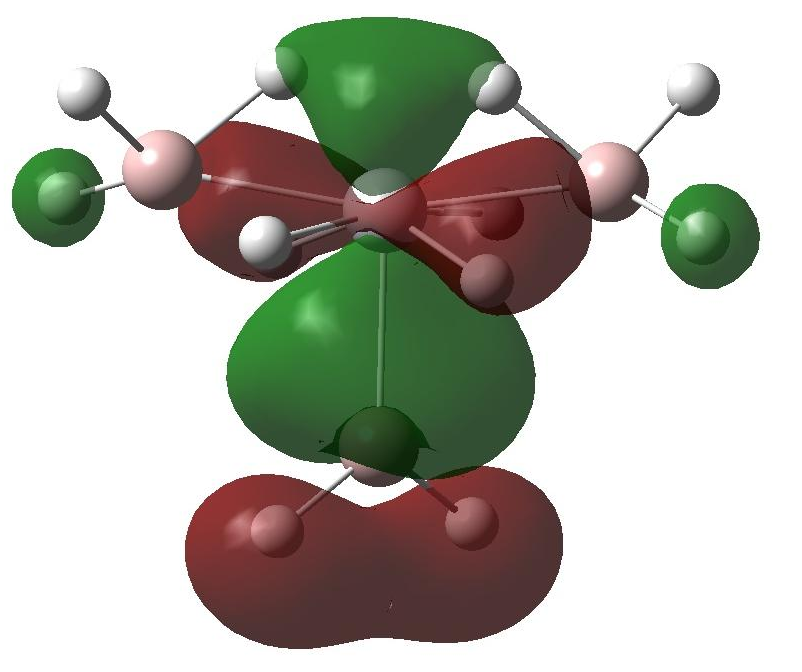
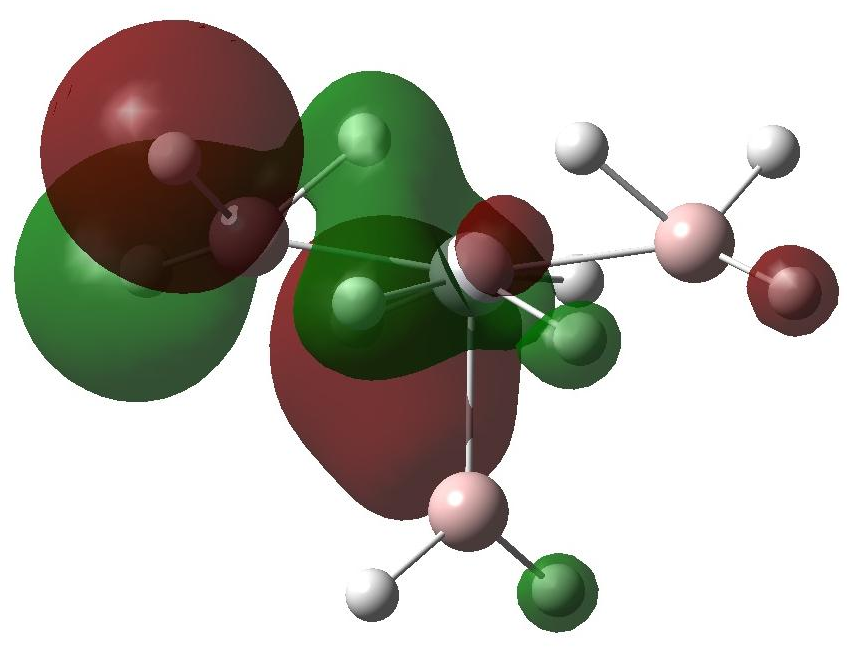
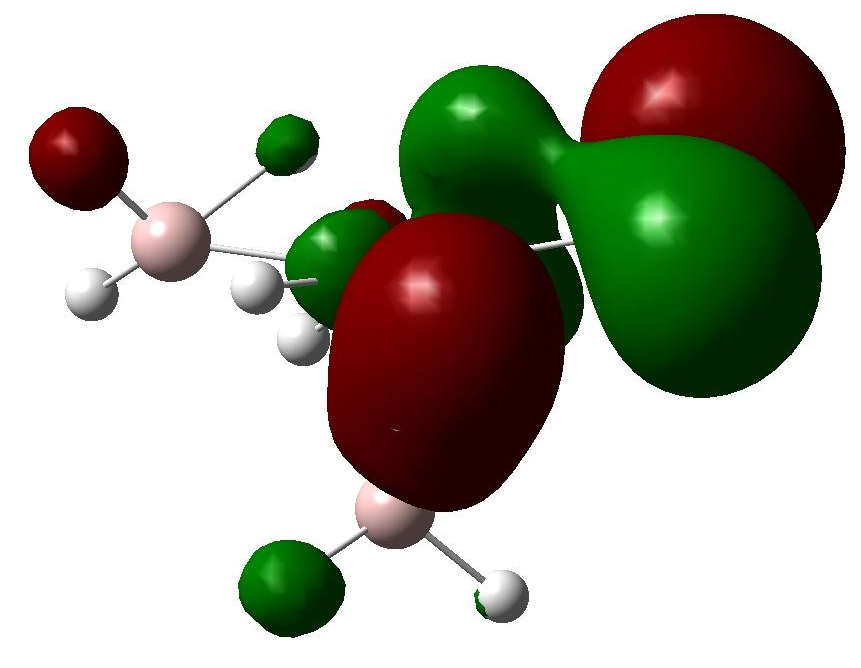
分子轨道和DOS的理论分析揭示了过渡金属的d轨道向Al-H和H-H的s*反键轨道的电子转移是储氢材料的热力学稳定性降低的关键因素,同时达到动力学速率增加的效果。
(3)氢气吸附反应
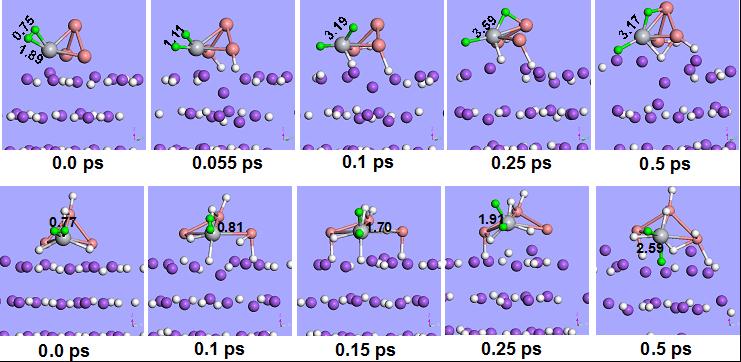
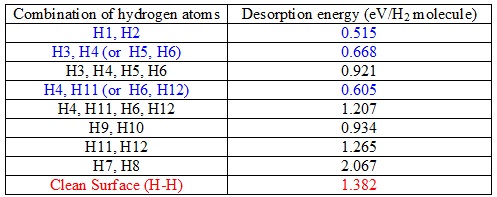
早期过渡金属与氢有较强相互作用,限制了氢溢流到载体中,从而导致过渡金属对氢解吸的催化能力,动力学模拟揭示了氢负离子转移有利于电子转移到过渡金属,这使过渡金属对氢解吸的催化效率显著提高。
(4)固态相变
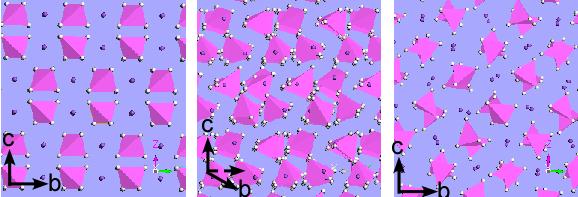
将Metadynamics应用在温度调控的常压相变模拟中,对NaAlH4晶体结构(I41/a)进行动力学研究,我们发现两个新的晶体结构(Cmcm, Pbcm),与拉曼光谱实验取得一致。

 沪公网安备 31010502006565号
沪公网安备 31010502006565号