
上海硅酸盐所在固态电解质配位结构设计取得系列进展
目前广泛使用的有机液态电解质存在易挥发、易燃易爆的安全隐患,设计具有高离子电导率的无机固态电解质是提高动力电池安全性的重要研究方向。然而,目前已有固体电解质存在离子电导率低、电化学窗口窄、界面结构不稳定的问题,限制它们的实际应用。最近,中国科学院上海硅酸盐研究所刘建军研究员带领的计算电化学团队在配位结构设计新型固态电解质材料方面取得了系列进展,相关成果先后发表在ACS Energy Lett.和J. Mater. Sci. Technol.期刊上。
就无机固态电解质材料而言,离子电导率主要取决于晶体结构中不同堆积多面体间连接结构,大部分具有八面体-四面体-八面体的迁移路径,由于锂离子占据高能量的四面体位点,导致高迁移势垒。通过 群体智能方法计算预测一类富锂层状氧化物Li3NbO4具有奇特的[NbO5]和[LiO5]五配位结构,形成了八面体-六面体-八面体的迁移路径,对应的迁移势垒(0.39eV)远低于岩盐结构I43m相Li3NbO4的迁移势垒(0.69eV),且具有4.3V的电化学窗口,进一步预测了具有本征空位缺陷的Li2Mg0.5NbO4也是一种高离子电导率的固体电解质。本研究为设计富锂层状固体电解质提供了一个新的策略。相关工作发表在ACS Energy Lett.(2021, 6, 3793-3800)。
此外,通过第一性原理分子动力学模拟研究了Li1+xAlxTi2-xP3O12(0≤x≤1)中的Li+离子扩散机制,发现Al3+掺杂将会通过电荷补偿引入间隙锂离子,被引入的间隙Li6a离子激活在Li1+xAlxTi2-xP3O12中相邻的6b位上的本征Li+离子,使Li+离子具有长程迁移特性;[PO4]四面体的畸变破坏了LiTi2P3O12中晶体场的高对称性,这种配位结构的调控改变了相邻的Li6b作用位点的势能以激发其活性,同时,具有适应结构变形能力的柔性[TiO6]多面体是Li1+xAlxTi2-xP3O12中Li+扩散的重要因素。相关工作发表在J. Mater. Sci. Technol.(2021, 73, 45-51)。
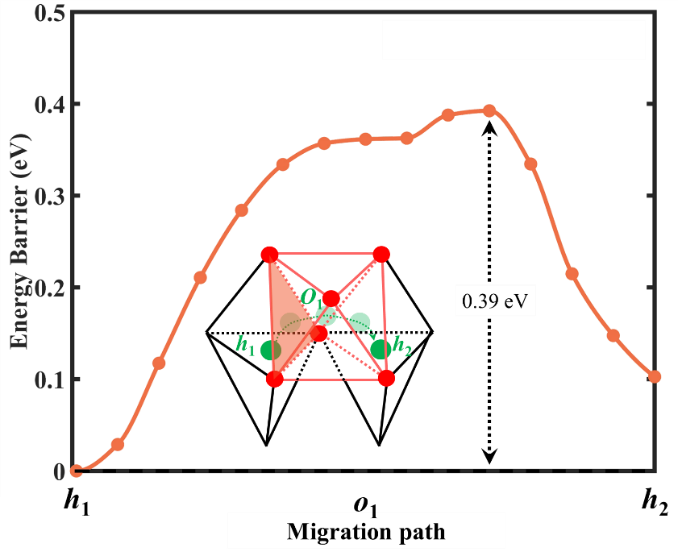
Li3NbO4化合物中Li+离子的迁移势垒(0.39 eV)远低于岩盐结构I43m相Li3NbO4的迁移势垒(0.69 eV)。
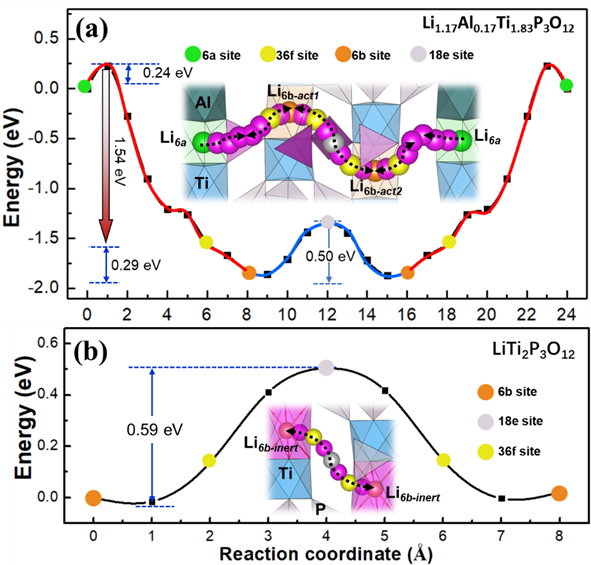
Al3+掺杂对Li+离子迁移势垒的调控关系:(a)Li1.17Al0.17Ti1.83P3O12中Li+离子迁移势垒;(b)LiTi2P3O12中Li+离子迁移势垒。
相关研究工作得到了国家自然科学基金委和上海科学技术委员会等项目的资助和支持。
附文章链接:
https://pubs.acs.org/doi/10.1021/acsenergylett.1c01781
https://linkinghub.elsevier.com/retrieve/pii/S1005030220308355

